MPS Disease
Last reviewed by adm_nyxl0f2y on January 12th, 2022.
What is MPS Disease?
MPS stands for mucopolysaccharidoses, which is a group of inherited lysosomal storage disorder. Lysosome plays an important role in the cell as it acts as the primary digestive unit. The enzymes in the lysosomes digest carbs, fats, and other nutrients.
What happens in people with MPS disease is that their cells are deficient in lysosomal enzymes. There is also a possibility that the enzymes in the lysosomes malfunction leading to excessive accumulation of complex carbohydrates like mucopolysaccharides in various parts of the body such as the eyes, skeleton, arteries, skin, teeth, ears, and other parts of the body.
Over time, it causes progressive cellular damage which will then affect the vital organs in the body. (1, 2, 3)

Image 1: An adult with an MPS disease.
Picture Source: i.ytimg.com
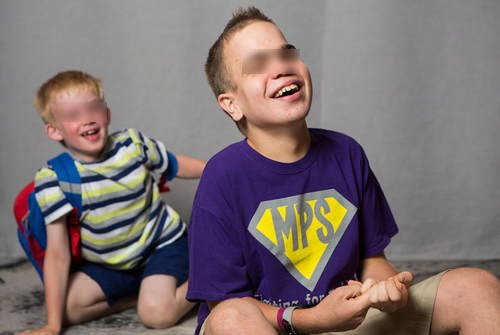
Picture 2: Children with MPS III disease.
Photo Source: mpssociety.org
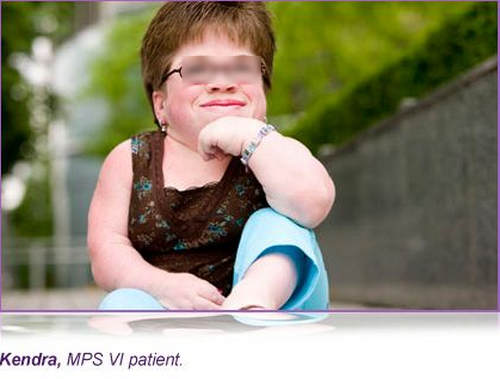
Photo 3: A classic manifestation of a person with MPS VI disease.
Image Source: i.pinimg.com

Image 4: A developmental stage of a person with MPS IV disease.
Picture Source: enablereview.biz
MPS disease is grouped into following:
MPS 1 H/S
Hurler-Scheie syndrome, it is a rare form of MPS and it pertains to people who have a less severe type of Hurler syndrome. However, it pertains to a more severe type than that of the Scheie syndrome. Clinical manifestations include short stature, a stiffness of the joints, coarse facial features, cardiac problems, clouding of the cornea, skeletal problems, and enlargement of the liver and spleen. A person has mild to moderate intellectual disability. The symptoms become evident when the person reaches 3-years-old. (2, 3)
MPS I H
Hurler syndrome, It is the severe form of MPS characterized by alpha-L-iduronidase deficiency. The symptoms start to appear as early as six months and become prominent as the infant transition into pre-school age. There is a noticeable developmental delay, respiratory tract infection, recurrent urinary tract infection, clouding of the eyes, deformed spine, a stiffness of the joints, and noticeably large tongue. (3, 4)
MPS II
Hunter syndrome, It is an inherited type of MPS. The symptoms start to appear when the person reaches the age of four. Clinical manifestations include developmental delays, a stiffness of the joints, short stature, coarse facial features, especially the lips, nostrils, and tongue, enlargement of the head, progressive hearing loss, broad chest, and short neck. (4, 5)
MPS III (A, B, C, and D)
Sanfilippo syndrome, symptoms include hyperactivity, developmental delays, sleep disorders, intellectual disability, and progressive hearing loss. There is also a possibility of seizure episodes, aggressive behavior, and unsteady gait. (4, 5)
MPS I S
Scheie syndrome, It is a mild form of MPS. A person lives a normal life but does have some issues with the stiffness of the joints, clouding of the cornea, aortic regurgitation, hearing difficulty, and carpal tunnel syndrome. The symptoms start to appear when the person reaches five years old.
MPS IV A and B
Morquio syndrome, it is caused by a deficiency of N-acetyl-galactosamine-6-sulfatase and beta-galactosidase enzymes. The symptoms are milder when compared to other subtypes of MPS disease. Classic manifestations include a prominent lower face, short neck, knock knees, abnormal side to side curvature of the spine, and prominent breast bones.
MPS VI
Maroteaux-Lamy syndrome, the symptoms vary from one person to another. Some people have mild symptoms while others have severe symptoms. The typical symptoms include coarse facial features, clouding of the cornea, skeletal problems, and umbilical hernia.
MPS VII
Sly syndrome, a person has a normal intelligence, although, some have a mild intellectual disability. Signs and symptoms include clouding of the cornea, short stature, skeletal problem, coarse facial features, cardiac-related diseases, and hernias. (3, 6, 7)
MPS IX
Hyaluronidase deficiency, a rare form of MPS characterized by short stature, cleft palate, recurrent ear infection, and occurrence of soft tissue masses.
What are the Symptoms of MPS?
Although MPS is a group of lysosomal enzyme disorders, people share similar symptoms which consist of the following:
- Multiple organ involvements
- Coarse facial features
- Bone and joint problems
- Short body built
- Breathing problems
- Enlargement of the liver and spleen
- Cardiac abnormalities
- Neurological problems
Infants usually appear normal at birth but the symptoms start to appear when the person reaches school age. In the case of MPS VII, nearly half of the pregnancy population are complicated by non-immune hydrops fetalis, which can be detected on routine ultrasound.
In some cases, the symptoms cannot be observed until the person reaches adolescence. Most cases of MPS are progressive in nature. A person’s physical and mental functions decline as he/she gets older, which could lead to life-threatening complications. (6, 8, 9, 10)
What is the Prevalence rate?
MPS disease happens one in every 25,000 births. However, some cases, particularly the mild forms go unrecognized or misdiagnosed. Hurler syndrome is more prominent in men than women.
Diagnosis
To diagnose MPS disease, the clinical manifestations will be thoroughly evaluated along with a series of tests. A urinalysis is ordered to check the level of mucopolysaccharides. An enzyme assay test will also be performed to check for a deficiency in lysosomal enzymes in the cells. A prenatal test and newborn screening test can also be performed, especially if the doctor is suspecting MPS disorders. (1, 3, 5)
MPS disease Treatment
- Naglazyme – It is approved by the US FDA for the treatment of MPS VI.
- Aldurazyme – It is used for the treatment of MPS I, specifically for patients with Hurler and Hurler-Scheie disorders. In fact, it is the very first approved treatment for MPS I.
- Elaprase – it is used to treat MPS II.
The treatment and management for MPS disease is yield towards the symptomatic relief. A healthcare team should work hand in hand to thoroughly and accurately manage the condition of the patient. For clinical manifestations like a hernia, skeletal problems, and carpal tunnel syndrome, a surgical approach might be necessary.
For severe clouding of the cornea, a corneal transplant might be necessary. For joint related problems, it can be improved using physical therapy and joint-specific exercises. Genetic counseling should be a part of the treatment and management of MPS.
Most importantly, the immediate family should be made aware of the patient’s condition so that they will know how to handle the patient’s condition. (1, 5, 9, 10)
References:
- https://rarediseases.org/rare-diseases/mucopolysaccharidoses/
- http://www.mpssociety.org.uk/diseases/
- https://www.mps1disease.com/en/patients/about/signs-and-symptoms.aspx
- https://www.medicinenet.com/mps_i_mucopolysaccharidosis_type_i_hurler_syndrome/article.htm
- https://en.wikipedia.org/wiki/Mucopolysaccharidosis
- http://oncofertility.northwestern.edu/resources/hurler-syndrome-mps-i
- http://jasonandjustinsjourneyx2.com/what-is-mps-2/
- https://raremark.com/sanfilippo-syndrome-mps-iii/articles/what-is-sanfilippo-syndrome-mps-iii–89
- https://radiopaedia.org/articles/mucopolysaccharidoses-2
- https://academic.oup.com/rheumatology/article/50/suppl_5/v4/1779302